
Scientific Conference: “Quality Management By Cloud Computing: Comprehensive Solution Through MasterControl Application.”
27 February, 2023
Mean Kinetic Temperature – MKT
9 March, 2023When medical device manufacturers consider placing devices on the market, they face the challenge of understanding the regulatory environment in different jurisdictions and the regulatory requirements that apply to them and their machines. As medical device regulators evolve their regulatory frameworks in a particular jurisdiction, they look for solutions to challenges they face from other countries and regions. Groups of countries in different parts of the world have looked to reduce barriers to trade by establishing a single market with standard regulatory requirements.
This article provides a comparison of the European Union (EU) 2017/745 Regulation1 and the Association of Southeast Asian Nations (ASEAN) Medical Device Directive (MDD). Of note, medical device directives and regulations are not addressed in this article and are not intended to serve as an interpretation for individual medical device manufacturers. Instead, this article is based on the analysis undertaken and presented at the 7th ASEAN Medical Device Committee Annual Meeting in Bangkok, Thailand, in December 2018.
Regional Comparison
The EU, which can trace its history to 1951, and ASEAN, which was formed in 1967, are organizations that bring together a number of countries to benefit their populations. They share essential similarities and have fundamental differences. The EU has 28 member states (pending Brexit), and the ASEAN has 10 member states. Both the EU and ASEAN were created to promote regional peace and prosperity through political and economic integration. Both organizations have committed to human rights: the EU through its Charter of Fundamental Rights and ASEAN through its Declaration of Human Rights. The EU and ASEAN are designed to integrate member nations’ economies into a single market, thereby promoting free trade.
Critical differences in the EU and ASEAN structure and mode of operation affect the regulatory frameworks for medical devices in the respective regions. Structurally, the EU member nations have conceded some degree of sovereignty to the EU, with mutual acceptance of regulatory decisions, whereas the ASEAN is structured as an intergovernmental organization. The EU has a parliament, while the ASEAN has an inter-parliamentary assembly that does not have authority over member nations. Several member states in the EU have adopted a common currency, whereas the ASEAN maintains separate currencies, preferring to encourage coordination and cooperation among finance ministers. The EU has 23 official languages, while the ASEAN has agreed to use English for meetings and correspondence.
The populations of the EU and ASEAN are comparable (636 million and 512 million, respectively). However, the size of the economies, as measured by gross domestic product, differ substantially, with the EU at $13 trillion and the ASEAN at $3 trillion (U.S. dollars). The average spending on healthcare per household is $2,673 for the EU and $107 for the ASEAN (U.S. dollars).
Evolution of Regulations for Medical Devices
An analysis of the regulations for medical devices in the EU and ASEAN reveals considerable similarities in both structure and content. This is no coincidence. Regulators from these regions interact and share experiences, best practices, and ideas, learning from each other’s work. Different areas update their regulatory framework at different times and in the context of their experience up to that point. Updates in one zone become informative to the other region. This interaction can be seen in the timeline shown in Figure 1 and is evidenced by matching text found in many parts of the ASEAN MDD and EU Regulation. As illustrated in Figure 1, the EU published new regulations for medical devices and in vitro diagnostics in 2017 and is transitioning to full implementation.
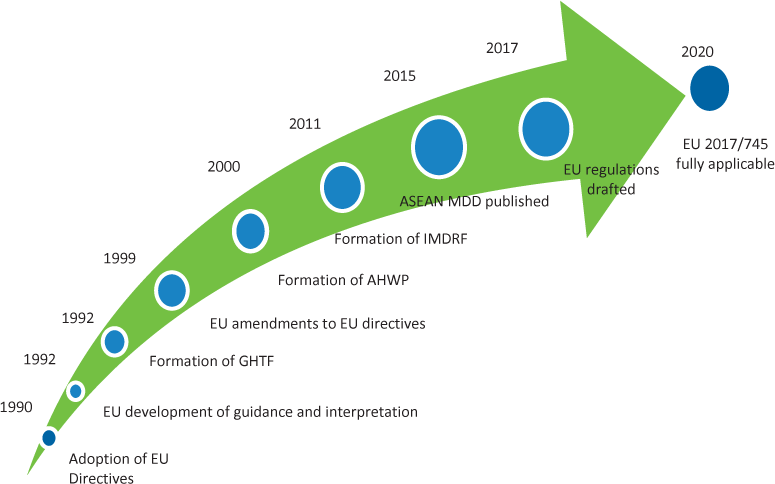
Figure 1. European Union (EU) and Association of Southeast Asian Nations (ASEAN) medical device directives and regulations: timeline of key milestones. Abbreviations used: AHWP, Asian Harmonization Working Party; GHTF, Global Harmonization Task Force; IMDRF, International Medical Device Regulators Forum; MDD, Medical Device Directive.
The Association for the Advancement of Medical Instrumentation (AAMI) advocates that regulators work regionally and globally. This work is resource intensive, and no country can afford to create and maintain the structure to regulate devices in today’s global marketplace. Furthermore, harmonizing standards is critical. Harmonization helps to provide market access for all manufacturers and increases the likelihood that safe and effective devices are accessible to patients.
Three key sources have influenced the development of regulations for medical devices:
- The Global Harmonization Task Force (GHTF) has encouraged a convergence in standards and regulatory practices related to medical device safety, performance, and quality and has promoted technological innovation and international trade.
- The International Medical Device Regulators Forum (IMDRF), which took over the GHTF’s mission in 2012, provides a means for discussing future directions in medical device regulatory harmonization.
- The Asian Harmonization Working Group studies and recommends ways to harmonize medical device regulations in Asia Pacific (including ASEAN) and other regions.
Directives and Regulations
Directives are legislative acts that set goals for member nations to achieve. Directives are not binding restrictions, and member nations decide how the directive is completed. As such, the ASEAN MDD allows each member country to execute provisions of the directive independently.
Harmonization helps to provide market access for all manufacturers and increases the likelihood that safe and effective devices are accessible to patients.
The EU 2017/745 is a regulation, an act binding upon all member countries of the union. This regulation supersedes the national laws of member nations.
Both directives and regulations have the impact of leading to greater harmonization across the respective region. Regulations accelerate this harmonization and address both the outcome and methods.
Essential Principles
The ASEAN and EU recognize that devices must achieve the performance intended by manufacturers, that they should be designed in a way that suits that purpose, and that they should be safe and perform as intended.
The ASEAN has developed the Common Submission Dossier Template, which requires manufacturers in all ASEAN jurisdictions to share the relevant Essential Principles (EPs) and methods used to demonstrate conformity.
The EU 2017/745 Regulation stipulates General Safety and Performance Requirements (GSPRs) that devices must meet; these can be used as inputs to the design and development process. The outputs of design and development can be verified and validated, as appropriate, against the GSPRs, thereby providing evidence and documentation to demonstrate compliance.
EPs and GSPRs have much in common; however, in some instances, GSPRs have additional detail or more specificity than EPs.
Collecting and sharing evidence regarding marketplace acceptance supports regulators and standards developers and ultimately enhances the safety and effectiveness of devices.
Role of Standards
International standards are building blocks for harmonized regulatory processes to ensure the safety, quality, and performance of medical devices. Regulations tell manufacturers what to do, while consensus standards tell them how to do it. A medical device manufacturer may use compliance with recognized standards to demonstrate compliance with the relevant EPs/GSPRs of medical devices. Both the EU 2017/745 Regulation and ASEAN MDD stipulate a role for standards as a voluntary means by which a manufacturer can demonstrate conformance with regulatory requirements. In the EU, if the manufacturer does not use harmonized standards, then they have to justify this technically, scientifically, and clinically to the notified body.
AAMI prefers global solutions, embracing the philosophy of “One standard, one test worldwide.” The association stresses openness, transparency, and due process, with representation from all stakeholders in standards development. Unless specific national conditions demand divergent national safety standards, they should be avoided. AAMI promotes safety and effectiveness of health technology, as well as access for care providers and patients. Standards promote regulatory, clinical, and patient acceptance of new technologies and reduce technical barriers to trade.
Standards supporting regulation across the medical device life cycle are important. Collecting and sharing evidence regarding marketplace acceptance supports regulators and standards developers and ultimately enhances the safety and effectiveness of devices.
Competent Authorities
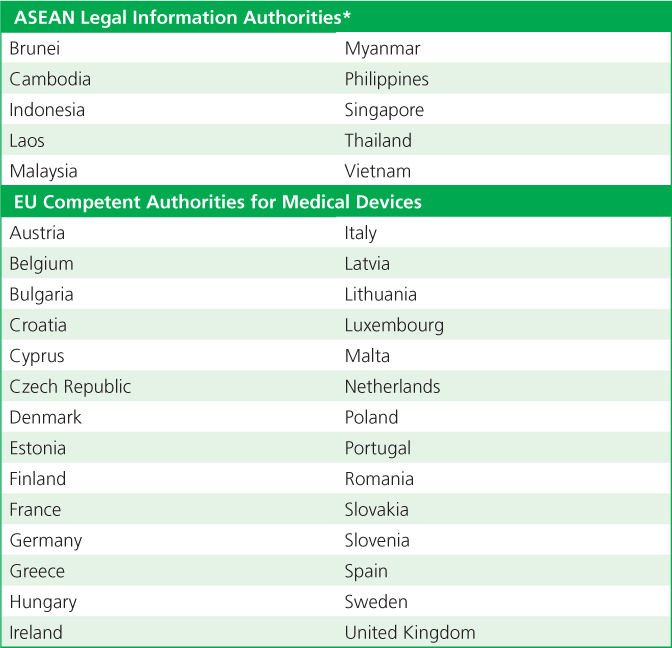
The ASEAN and EU require member states/countries to select and identify Legal Information Authorities and Competent Authorities for Medical Devices (CAMDs), respectively (Table 1). This authority usually is the individual member state’s ministry of health or an agency connected to the ministry of health. In the EU, the European Commission is responsible for properly executing regulations and for coordinating the activities of the member states.
Table 1. Member countries/states Association of Southeast Asian Nations (ASEAN) Legal Information Authorities and European Union (EU) Competent Authorities for Medical Devices.
Stakeholders
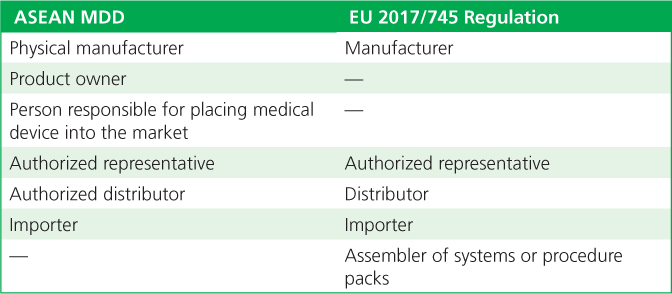
The ASEAN and EU recognize similar medical device parties in the supply chain. Both entities recognize the importance of organizations involved in the life cycle of a device—starting with its design and moving through manufacturing, distribution to the user, and monitoring for safety and performance. Although they use somewhat different terminology, the ASEAN and EU recognize the pivotal role and responsibilities of the organization whose name is on the product. This organization has overall responsibility for the product’s life cycle, whether it performs the activities itself or has other organizations undertake some or all of the activities on its behalf (Table 2).
Table 2. The terminology used for parties in the supply chain in the Association of Southeast Asian Nations (ASEAN) Medical Device Directive (MDD) and European Union (EU) 2017/745 Regulation.
Defining Medical Devices
The ASEAN MDD and EU 2017/745 Regulation define medical devices similarly, drawing from past EU directives and GHTF document versions. Standards from the International Organization for Standardization and AAMI also use the same sources for defining medical devices. The definitions capture the purposes of diagnosis, prevention, monitoring, treatment, and alleviation of disease or compensation for an injury; investigation, replacement, or modification of the anatomy or physiological processes; or control conception.
The EU 2017/745 Regulation definition includes, for the first time, equipment for sterilizing medical devices as devices in their own right. Before this, sterilization devices fell outside of the definition of medical devices. With this change, sterilization equipment falls into the category of a medical devices rather than accessories under the EU Medical Device Regulation.
Items and equipment with features that are characteristic of medical devices but don’t have specific medical purposes, having only cosmetic purposes (e.g., liposuction equipment, nonprescription contact lenses), are listed in an annex in the EU 2017/745 Regulation and regulated as if they are medical devices.
References
Brad Schoener; Eamonn Hoxey
Biomed Instrum Technol (2019) 53 (3): 208–213.
https://doi.org/10.2345/0899-8205-53.3.208


