Comparator Management (P.2)
28 March, 2022
QRM Application in GMP
17 April, 2022This is one in a series of guidances that provides recommendations for applicants preparing the
Common Technical Document for the Registration of Pharmaceuticals for Human Use (CTD). This guidance presents the agreed-upon common format for the preparation of a well-structured harmonized application that will be submitted to regulatory authorities. A common format for the technical documentation will significantly reduce the time and resources used to compile applications for the registration of human pharmaceuticals and will ease the preparation of electronic submissions. Regulatory reviews and communication with the applicant will be facilitated by a standard document of common elements. In addition, exchange of regulatory information among regulatory authorities will be simplified.
Guidance for industry on preparing the CTD has been divided into four guidance documents on (1) the organization of the CTD, (2) the quality section (3) the efficacy section (4) the safety section. For specific information on the quality, efficacy, and/or safety sections of the CTD, see the individual guidances for an industry that discuss those parts of the CTD. For general information about submitting a marketing application in the CTD format in the U.S. region, as well as specific information about Module 1 (U.S. administrative information), see the guidance
for industry, General Considerations for Submitting Marketing Applications According to the
ICH/CTD Format.2 The CTD guidances are intended to be used together with other ICH and
Agency guidances. Please refer to those guidances for detailed information about the contents of an application.
1. The CTD
Through the ICH process, considerable harmonization has been achieved among the three regions (Japan, Europe, and the United States) in the technical requirements for the registration of pharmaceuticals for human use. However, until now, there has been no harmonization of the organization of a submission. Each region has its own requirements for the organization of the technical reports in the submission and for the preparation of the summaries and tables. In Japan, the applicants must prepare the GAIYO, which organizes and presents a summary of the technical information. In Europe, expert reports and tabulated summaries are required, and written summaries are recommended. The U.S. FDA has guidance regarding the format and content of the new drug application submission. To avoid generating and compiling different registration dossiers, this guidance describes a harmonized format for the CTD that will be acceptable in all three regions.
2. Preparing and Organizing the CTD
This guidance addresses the general organization of the information to be presented in
the CTD application for new pharmaceuticals (including biotechnology-derived products). Guidance documents also are available that discuss the Quality, Efficacy, and Safety sections of the CTD. These guidances are not intended to indicate what studies are required. The guidances merely indicate an appropriate format for the data that have been acquired. Applicants should not modify the overall organization of the CTD. However, in the Nonclinical and Clinical Summaries sections of the CTD, applicants can modify individual formats to provide the best possible presentation of the technical information to facilitate the understanding and evaluation of the results. Throughout the CTD, the display of information should be unambiguous and transparent, to facilitate the review of the basic data and to help a reviewer become quickly oriented to the application contents. Text and tables should be prepared using margins that allow the document to be printed on both A4 paper (E.U. and Japan) and 8.5 x 11” paper (U.S.). The left-hand
margin should be sufficiently large that information is not obscured through binding. Font sizes for text and tables should be of a style and size that are large enough to be easily legible, even after photocopying. Times New Roman, 12-point font is recommended for narrative text. Acronyms and abbreviations should be defined the first time they are used in each module.
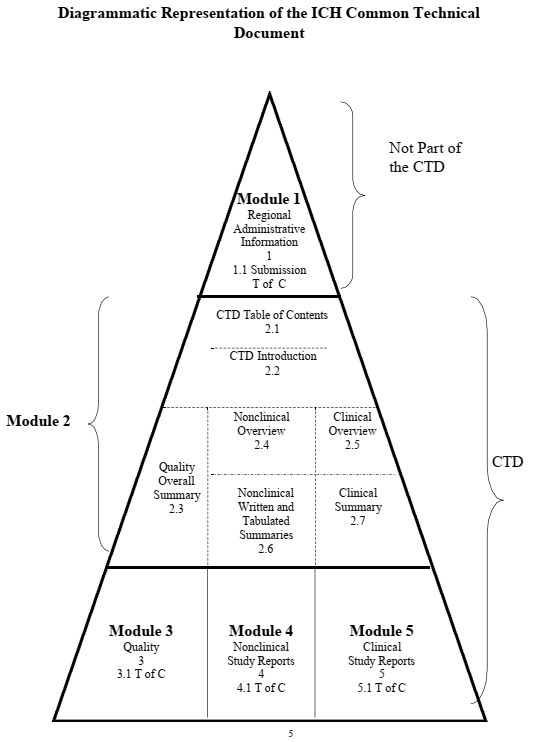
3. Structure
Module 1 is region-specific. Modules 2, 3, 4, and 5 are intended to be common for all regions. Conformance with the CTD guidances should help ensure that these four modules are provided in a format acceptable to the regulatory authorities.
- Module 1. Administrative Information and Prescribing Information
This module should contain documents specific to each region, for example, application forms or the proposed label for use in the region. The content and format of this module can be specified by the relevant regulatory authorities. For information about this module see the guidance for industry, General Considerations for Submitting Marketing Applications According to the ICH/CTD Format.
- Module 2. Common Technical Document Summaries
Module 2 should begin with a general introduction to the pharmaceutical, including its
pharmacologic class, mode of action, and proposed clinical use. In general, the
introduction should not exceed one page.
Module 2 should contain 7 sections in the following order:
· CTD Table of Contents
· CTD Introduction
· Quality Overall Summary
· Nonclinical Overview
· Clinical Overview
· Nonclinical Written and Tabulated Summaries
· Clinical Summary.
Because Module 2 contains information from the Quality, Efficacy, and Safety sections of the CTD, the organization of the individual Module 2 summaries is discussed in three separate documents:
· M4Q: The CTD — Quality
· M4S: The CTD — Safety
· M4E: The CTD — Efficacy.
- Module 3. Quality
Information on Quality should be presented in the structured format described in the guidance, M4Q.
- Module 4. Nonclinical Study Reports
The Nonclinical Study Reports should be presented in the order described in the guidance
M4S.
- Module 5. Clinical Study Reports
The human study reports and related information should be presented in the order described in the guidance M4E.